

بتا تالاسمی و آلفا تالاسمی از بیماری های شایعی هستند که هموگلوبین را درگیر میکنند. در بدن انسان گلبول های قرمز وظیفه نقل و انتقال اکسیژن را بر عهده دارند و نقش اصلی را در متابولیسم بدن بازی میکنند. ماده اصلی که در گلبول های قرمز وجود دارد و درواقع ابزار انتقال گازهای تنفسی است هموگلوبین نام دارد. تالاسمی نامی است که به گروه عمده ای از اختلالات هموگلوبین اطلاق میشود که خود به دو گروه آلفا تالاسمی و بتا تالاسمی تقسمی میشوند.
پیشتر در مقاله ای جداگانه به آلفا تالاسمی و مقدماتی در مورد هموگلوبین و نحوه ایجاد تالاسمی پرداختیم که میتوانید با کلیک بر این لینک به آن مقاله بروید. در این مقاله به انواع بتا تالاسمی، نحوه ایجاد، علائم و درمان آن میپردازیم.
بتا تالاسمی چیست؟
همچنانکه اشاره شد، همگلوبین طبیعی دو زنجیره آلفا و دو زنجیره بتا دارد که از نسبت ۱:۱برخوردارند. برای ساخت دو زنجیره بتا نیاز به دو ژن است که یکی از مادر و دیگری از پدر به ارث می رسند. فقدان از هر کدام از این ژنها علایم خاص خود را دارد و نوع متفاوتی از تالاسمی بتا ایجاد می کند. در حالت عادی برای تولید زنجیره بتا کامل باید mRNA نسخه برداری شده از روی ژن آن بر روی کروموزوم ۱۱ کاملا ترجمه شود و اگر ترجمه در هر جایی از mRNAقطع شود منجر به ساخت زنجیره بتا کوتاهتری خواهد شد.
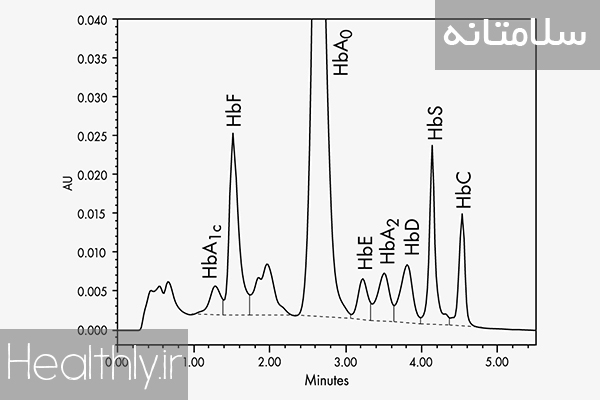
انواع تالاسمی بتا
الف) تالاسمی مینور(+β)
اگر توقف ترجمه در انتهای زنجیره بتا رخ دهد و زنجیره نزدیک به اتمام باشد که به تالاسمی مینور یا تالاسمی صفت (+β) موسوم است. یعنی فرد فقط صفت تالاسمی را دارد و از نظر بالینی فاقد علامت است و یا مشکل خاصی ندارد. بیمار در آزمایشات روتین و یا هنگام چک آپ بدن، دارای آنمی هیپوکروم میکروسیتک است. بیمار کم خونی خفیفی دارد و هماتوکریت آن ۳۰تا ۳۳درصد و Hbآن ۹تا ۱۱گرم در دسی لیتر است. اغلب MCVکمتر از ۷۵دارند. در الکتروفورز ویژگی های این افراد به صورت زیر است:
HbA=94-95% HbA2=3.5-7.5% HbF=1-3%
هر چه پلی پپتید ترجمه شده کوتاهتر باشد، شدت آنمی بیشتر و میزان HbF بالاتر است. پس میزان HbA2 در این افراد بالاست. HbA2 نرمال بین ۱/۵تا ۳/۵ درصد است. در لام خون محیطی افراد دچار تالاسمی مینور، آنمی هیپوکروم میکروسیتک با target cellوجود دارد (چون مقدار هموگلوبین درون RBCدر تالاسمی مینور کم است.) بر این اساس به عنوان یک سازگاری فیزیولوژیک، برای اکسیژن رسانی مطلوب تعداد RBCها افزایش می یابد و از ۵تا ۵/۵میلیون در هر میکرولیتر بالاتر می رود. از طرف دیگر، مقدار HbA2در خون نرمال است.
نکته شایان ذکر در مورد افراد با تالاسمی مینور این است که در صورت وقوع همزمان آنمی فقرآهن (بخصوص در زنان جوان با هایپرمنوره یا در موقع حاملگی) مشکل جدی تر می شود. در حالت عادی تالاسمی مینور نیاز به درمان تجربی با آهن ندارد و حتی تجویز اهن مشکل ساز می شود چون بدلیل لیز بیشتر RBC ها و ترن آور سلولی بیشتر فرد دچار افزایش بار آهن می شود. فقط در صورتی باید اهن مصرف نمود که فریتین فرد کاهش نشان دهد.
ب)تالاسمی ماژور (β۰β۰)
در این افراد زنجیره هموگلوبین کوهتاهتری نسبت به تالاسمی مینور ساخته می شود که به طور کامل رسوب می کند و در گلبولها ی قرمز اجسام هینز (Heinz body) بوجود می آید و لیز شدیدتری رخ می دهد. این افراد در دوره جنینی و ابتدای تولد مشکل ندارند چون هموگلوبین جنینی (αα/γγ) (HbF) دارند ولی چون در اواخر بارداری به طور طبیعی بایستی زنجیره های دلتا جای خود را به زنجیره های بـتا بدهند تا هموگلوبین HbAساخته شود، مشکلات بروز می کنند. بطوریکه از ۶ماهگی به بعد، شیر خوار از شیرخوردن امتناع نموده و افزایش وزن ندارد طحال وی نیز ممکن است قابل لمس باشد. این افراد کم کم دچار آنمی شده و برای بقا بایستی تا پایان عمر خون دریافت کنند. علایم آزمایشگاهی بتا تالاسمی ماژور عبارتند از: آنمی شدید، هموگلوبین بین ۶تا MCV ،۷پایین، HbFیا HbA2 و یا هر دو بالا، Target cellزیاد در لام خون محیطی. در الکتروفورز هموگلوبین خون فرد مبتلا به تالاسمی ماژور:
HbA=low (NL.=97%) HbA2=5-10% (NL=5-10%) HbF=60-70% (NL=1-3%)
در برخی بیماران HbFممکن است تا %۹۰ افزایش یابد و از نظر بالینی شبیه به HbFHمی شوند که فاقد علامت بالینی
هستند. در معاینه بالینی بیماران مبتلا به تالاسمی ماژور به دلیل لیز زیاد گلبولهای قرمز زردی و اسپلینومگالی دارند و به دلیل خون سازی در مغز استخوان، استخوانهای پهن و استخوانهای جمجمه آنها بزرگ می شوند. سینوس ها و استخوان پتروس آنها نیز بزرگ است. پیشانی و گونه های برجسته دارند و صورت آنها حالت موش خرما (Chipmunk face) به خود می گیرد. استخوانهای مذکور نازک و مستعد شکستگی پاتولوژیک می شوند. طحال و کبد افراد مبتلا به دلیل خونسازی اکسترامدولاری بزرگ شده و زخم کف پا و سنگ پیگمانته کیسه صفرا ایجاد می شود. عقب ماندگی رشد شدید و CHF با برون ده بالا نیز دارند.
درمان این بیماران شامل تزریق خون مکرر (برای سرکوب مغز استخوان و کاهش یا توقف ساخت هموگلوبین معیوب)، مکمل اسید فولیک، دسفرال (برای خروج اهن اضافی از بدن) و در نهایت پیوند مغز استخوان آلوژنیک (به عنوان درمان قطعی) است. توصیه شده است که پیوند مغز استخوان در اوایل کودکی و قبل از آسیب بافتی انجام شود. در ۸۰تا ۹۰ درصد موارد درمان قطعی است. ژن درمانی در این مورد مفید نبوده است.
هر واحد خون، ۲۵۰تا ۳۰۰ میلی گرم آهن دارد و ۲واحد خون معادل مصرف یک تا دو سال آهن خوراکی است. اگر فردی بیش از ۱۰۰واحد خون دریافت نماید با توجه به دفع کند آهن از بدن، ممکن است فرد به تجمع بیش از حد آهن یا هموسیدروزیس مبتلا شود که علایم آن شبیه هموکروماتوز یعنی افزایش سطح فریتین، GTTمختل، تاخیر در بلوغ، سیروز کبدی و کاردیومیوپاتی می باشد. تشخیص هموسیدروزیس با بیوپسی کبد و یا روش غیر تهاجمی اسکن SQUID1است. برای درمان هموسیدروزیس هم از شلاترهای آهن مثل دسفرال (تزریقی) و دفراسیروکس (خوارکی) استفاده می شود. این داروها عوارض LFTبالا و افزایش مختصر کراتینین را دارند.
بتا تالاسمی اینتر مدیا (+β۰β)
در این حالت طول زنجیره هموگلوبین ساخته شده حد واسط دو نوع مینور و ماژور است. بیمار در کودکی هیچ علامتی ندارد ولی در هنگام بلوغ علامت دار می شود. برخی مواقع نیاز به انتقال خون دارند و بدون انتقال خون زیاد، طول عمر زیادی خواهند داشت. در موارد شدید آنمی، اسپلینوکتومی می شوند. درمان انها مشکل است چون کم خونی آنها به دلایل متعدد و معمول همچون عفونت، آغاز بلوغ، اسپلینومگالی و هایپراسپلنیسم تشدید می شود. برای درمان این بیماران، تزریق خون، تجویز هیدروکسی اوره و سیتارابین مفید است. این دو دارو باعث تحریک سنتز HbF می شود. در صورت تزریق خون بایستی به پیامدهای آن به خصوص هموسیدروز توجه داشت.
تالاسمی بتا و ازدواج
تاکید می شود که در تالاسمی نسبت زنجیره های آلفا به بتا مهم است. این نسبت در حالت نرمال بین ۰/۷تا ۱ است. اگر بیشتر از ۱/۲شود یعنی زنجیره آلفا زیادتر است و رسوب می کند و فرد بتا تالاسمی دارد. و اگر کمتر از ۰/۷باشد فرد مبتلا به آلفا تالاسمی است. چون زنجیره بتا بیشتر است.
اگر فرد مبتلا به تالاسمی بتا مینور با یک فرد مبتلا به تالاسمی آلفا trait ازدواج کند فرزند آنها مبتلا به “آلفا-بتا تالاسمی” می شود که وضعیت بهتری است چون بین زنجیره های آلفا و بتا نسبت نزدیکی برقرار است. فرد یک آنمی خفیف بدون همولیز دارد و الکتروفورز هموگلوبین هم طبیعی است.
اگر فرد مبتلا به تالاسمی آلفا با فرد مبتلا به کم خونی سلول داسی شکل ازدواج کند، فرزند آنها دچار “سایکل آلفا تالاسمی” خواهد بود. این فرزند نسبت به فرد دچار کم خونی داسی شکل هموزیگوت وضعیت بهتری دارد چون زنجیره بتا اضافی رسوب می کند و مانع داسی شکل شدن گلبول ها می شود.
تالاسمی ماژور فقط در یک حالت رخ می دهد و آن هم زمانی است که هر دو والد β۰ باشند. نوزاد مبتلا تا مدتی ممکن است زنده بماند.