
آلفا تالاسمی و بتا تالاسمی از بیماری های شایعی هستند که هموگلوبین را درگیر میکنند. در بدن انساس گلبول های قرمز وظیفه نقل و انتقال اکسیژن را بر عهده دارند و نقش اصلی را در متابولیسم بدن بازی میکنند. ماده اصلی که در گلبول های قرمز وجود دارد و درواقع ابزار انتقال گازهای تنفسی است هموگلوبین نام دارد. تالاسمی نامی است که به گروه عمده ای از اختلالات هموگلوبین اطلاق میشود که خود به دو گروه آلفا تالاسمی و بتا تالاسمی تقسمی میشوند.
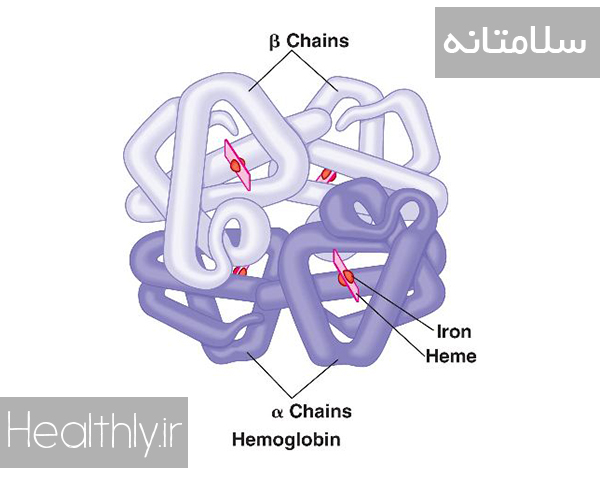
هموگلوبین چیست؟
هموگلوبین مولکولی است که از چهار زنجیره پپتیدی تشکیل شده است. هر زنجیره دارای یک Heme است که یک حلقه پروتوپورفیرین- ۹ حاوی اهن فریک یا + Fe2 است. هر جزء Heme به یک مولکول اکسیژن متصل می شود. لذا، هر مولکول هموگلوبین توان حمل چهار مولکول اکسیژن را دارد (توجه شود که در هر گلبول قرمز حدود ۲۷۰میلیون مولکول هموگلوبین وجود دارد) ژنهای کد کننده هموگلوبین روی کروموزوم های ۱۱و ۱۶قرار دارند. هر زنجیره پپتیدی هموگلوبین میتواند از نوع ،α β، δدلتا و γگاما باشد. ژن زنجیره αروی کروموزوم ۱۶هست و لی ژن بقیه زنجیره ها روی کروموزوم ۱۱قرار دارد. هر فرد حتما دو زنجیره آلفا و دو زنجیره دیگر از هر یک از سه نوع دیگر دارد. به این ترتیب با ترکیب ۲ زنجیره آلفا و دو زنجیره دیگر از نوع مذکور انواعی از همگلوبین شکل می گیرد که اسامی خاص دارند.
تالاسمی چیست؟
به طور کلی تالاسمی یا اکتسابی است و یا ارثی (ژنتیکی.) در این جا در مورد انواع ژنتیکی بحث می کنیم. برای اینکه هموگلوبین داخل گلبول قرمز محلول و به صورت مایع باشد باید مقدار زنجیره آلفا و بتا از نظر وزنی و تعدادی در تعادل ۱:۱ باشند. هر شرایطی که منجر به انحراف در تعادل فوق شود، موجب زیادی یک زنجیره خواهد شد که زنجیره اضافی در داخل سلول رسوب می کند. رسوب هر یک از این زنجیره های پپتیدی برای سلول سمی است.
تالاسمی ها بر اساس اینکه زنجیره آلفا و یا بتا زیادتر از دیگری باشد به دو نوع کلی زیر تقسیم می شوند:
آلفا تالاسمی: در این حالت زنجیره (های) آلفا نقص دارند و زنجیره های بتا اضافی تولید می شوند لذا در سلول رسوب می کنند. این حالت کمتر برای گلبول قرمز سمی است و موجب مرگ آن نمی شود
بتا تالاسمی: در این حالت زنجیره (های) بتا نقص دارند و زنجیره های آلفا اضافی تولید می شوند لذا در سلول رسوب می کنند. این حالت برای گلبول قرمز بسیار سمی است و موجب مرگ آن در داخل مغز استخوان می شود.
هر یک از انواع تالاسمی آلفا و بتا بر مبنای وضعیت ژنتیکی فرد به چند دسته تقسیم می شوند که علایم بالینی و آزمایشگاهی خاصی را دارند.
انواع تالاسمی آلفا
همچنانکه ذکر شد، ژن زنجیره α روی کروموزوم ۱۶ قرار دارد. برای ساخت دو زنجیره آلفا نیاز به چهار ژن است که دو تا از مادر و دو تا از پدر به ارث می رسند. چون احتمال حذف دو ژن کمتر از یک ژن است لذا احتمال تالاسمی آلفا کمتر از تالاسمی بتا، ولی در مقابل خطرناکتر از آن است. تالاسمی های آلفا دو دسته اند. یک دسته به حذف کامل ژن مربوط می شوند و لذا شامل چهار نوع حذف ۳ ،۲ ،۱ و یا ۴ ژن می باشند. انواع دیگر با حذف ژن همراه نمی باشند بلکه مربوط به بیان ناکامل ژن هستند. در اینجا به چهار نوع تالاسمی مربوط به حذف ژن پرداخته می شود.(که شایع تر هستند).
الف)آلفا تالاسمی با وضعیت ناقل خاموش (Silent career)
در این نوع آلفا تالاسمی فقط یکی از چهار جایگاه ژنی مربوط به ساخت هموگلوبین حذف شده است و سه جایگاه دیگر فعال هستند. تالاسمی خاموش، شایع ترین شکل بیماری تالاسمی در کشورهای آسیایی است. این افراد مشکل بالینی دارند. در این افراد مقدار % ،HbA=97 و مقدار Hb توتال، HbF و ،HbA2 الکتروفورز، آهن و فریتین خون همه نرمال هستند لذا جز با بررسی ژنتیکی، قابل تشخیص از افراد عادی نمی باشند. تنها مشکل مربوط به این افراد، هنگام ازدواج دو فرد با تالاسمی خاموش پیش می آید که با توجه به احتمال به ارث رسیدن همزمان دو کروموزوم فاقد ژن کد کننده زنجیره آلفا، درصدی از فرزندان آنها فاقد دو ژن و در نتیجه مبتلا به شکل شدید تری از تالاسمی خواهند بود. یک نکته قابل توجه در مورد این افراد، فراوانی بیشتر آنها در مناطق اندمیک مالاریا است. افراد مبتلا به تالاسمی خاموش، به مالاریای ناشی از پلاسمودیم ویواکس (فرم کم خطرتر از پلاسمودیوم فالسیپاروم) حساسیت بیشتری دارند و گلبول های قرمز آنها سریعتر لیز شده و عامل بیماری در خون آنها در معرض سیستم ایمنی قرار می گیرد و احتمال حذف ان و طول عمر فرد بیشتر است.
ب)آلفا تالاسمی صفت (Trait)
به وضعیتی از تالاسمی اطلاق می شود که فرد دو ژن از ۴ ژن کد کننده زنجیره های آلفا را ندارد. به طور طبیعی این دو ژن ممکن است مربوط به یک والد(–/αα) و یا مربوط به دو والد باشند (-α-/α) باشند. که به ترتیب حالت های سیس و ترانس نام دارند. وضعیت ژنتیکی (–/αα) ایجاد صفت آلفا-تالاسمی- ۱ می کند که هر دو ژن معیوب از یک والد گرفته شده اند و در نسل بعد خطر بیشتر دارد.
اما، ژنوتیپ (-α/-α) موجب صفت آلفا تالاسمی- ۲ می نماید که در آن یک ژن معیوب از هر والد گرفته شده است. صفت آلفا تالاسمی- ۱ هم ناقل خاموش است ولی صفت آلفا تالاسمی- ۲ شبیه تالاسمی بتا مینور است. این افراد آنمی هیپوکروم میکروسیتک با همگلوبین ۱۲تا ۱۳و MCV=70-80flو RBC بالا دارند. از نظر تشخیصی، در این افراد HbAپایین ولی HbA2 بالاست و فرد آنمی فقر آهن ندارد. در کل، در الکتروفورز آلفا تالاسمی صفت، مقادیر زیر تشخیصی هستند:
Hb=12-13 mg/dL HbA=85-90% HbA2=1-2% HbF=0-1%
ج)تالاسمی هموگلوبین H
اگر در فردی سه ژن کد کننده رنجیره هموگلوبین آلفا وجود نداشته باشد، بیماری هموگلوبین Hنام دارد. حذف همزمان سه ژن (—/α) موجب تولید هموگلوبین HbH می شود. در این افراد هر چهار زنجیره بتا است (β۴) و لذا نسبت آلفا به بتا بسیار کم است لذا در لام خون محیطی درون RBC ها انکلوزیون های ناشی از رسوب بتا وجود دارد و به انها نمای توپ گلف می دهد. این افراد آنمی شدیدتری دارند و Hbآنها بین ۶تا ۱۰گرم در دسی لیتر و HbA ،MCH=60-70بین ۷۰تا ۹۰درصد و HbH بین ۵تا ۳۰درصد دارند. تشخیص مهم آنها با مشاهده انکوزیون های رسوب بتا در RBCهای خون محیطی است. این افراد علایم شبیه به تالاسمی بتا نوع اینترمدیا دارند. در مجموع این نوع تالامسی هم با حیات منافات ندارد و در دوره کودکی علایم بالینی ندارند ولی از نوجوانی به بعد علایم به صورت آنمی و اسپلینومگالی در آنها دیده می شود. بعضا ممکن است در شرایطی نیاز به تزریق خون داشته باشند. به عنوان مثال در خانم های باردار موقع زایمان ممکن است خون لازم داشته باشند. این افراد ورزشکار حرفه ای هم نمیت وانند باشند. شانس بقای آنها تا میانسالی هم وجود دراد. برخی موارد اسپلینومگالی هم برای آنها مفید است. این افراد بایستی از مصرف داروهای اکسیداتیو خودداری نمایند. در الکتروفورز اختصاصی وجود HbH (تترامرهایβ۴) در آنها تشخیصی است. در این بیماران HBA2 و HbF بالا نمی باشد.
بیشتر بخوانید: MCH در آزمایش
د) هیدروپس فتالیس یا هموگلوبین بارت (Hb Baret)
در این افراد هیچ یک از چهار ژن کد کننده زنجیره آلفا هموگلوبین وجود ندارند و زنجیره بتا هم ساخته نمیشود حدود ۵ تا HbH % 10دارند و HbA=0است. لذا هموگلوبین بارت یا γ۴ تولید می شود. هموگلوبین بارت با حیات منافات دارد. این هموگلوبین γ۴در مقایسه با سایر واریانت های هموگلوبین بیشترین میل ترکیبی را با اکسیژن دارد لذا اکسیژن را در بافت ها رها نمی کند و در دوره جنینی باعث هیپوکسی شدید می شود. در پاسخ به این وضعیت جینن شروع به اکسترامدولاری هماتوپوئیزیس می کند که نتیجه آن بزرگ شدن طحال، پهن شده استخوانهای صورت، نارسایی قلب و بروز ادم جنرالیزه در بدن جنین است. این جنین ها یا اغلب مرده بدنیا می آیند و یا مدت کوتاهی پس از تولد می میرند. پس هیچوقت آدم بالغ با هموگلوبین بارت دیده نمی شود.
علائم و درمان آلفا تالاسمی
همانطور که گفته شد در ۴ نوع آلفا تالاسمی که وجود دارد در آلفا تالاسمی صفت ۲ و هموگلوبین H علائم کم خونی وجود دارد و در مواقع ضروری ممکن است نیاز به انتقال خون پیدا کنند؛ و آلفا تالاسمی هموگلوبین بارت که کلا نوزاد به دنیا نمی آید و یا بعد از تولد میمیرد، سایر انواع آلفا تالاسمی علائم اختصاصی و شدیدی ندارند و به طبع آن درمان های اختصاصی نیز ندارند. در واقع اهمین بررسی انواع آلفا تالاسمی در حیطه ازدواج مشخص میشود که هنگامی دو شخص که مشکلات خونی دارند بخواهند با یک دیگر ازدواج کنند.
به طور کلی ازدواج فرد داراي آلفا تالاسمی مینور با شخص بتا تالاسمی مینور نه تنها مشکل ساز نیست بلکه در صورت بچه دار شدن آن ها فرزندشان داراي Hb هاي متعادل تري نسبت به هر یک از والدین خود میباشد. ینی در واقع اگر فردي داراي آلفا تالاسمی مینور باشد ازدواج او با یک فرد بتا تالاسمی مینور نسبت به یک فرد سالم ارجحیت دارد زیرا در این صورت فرزندان آنها سالم تر خواهند بود و عدد هموگلوبین بالاتري خواهد داشت اما اگر ژن معیوب در هر دو نفر یکسان باشد مثلا هر دو بتا تالاسمی مینور باشند ممکن است بچه هاي آن ها مبتلا به بتا تالاسمی ماژور شوند (احتمال %۲۵)
سلام وقت بخیر اگر هردو نفر آلفا تالاسمی ماینور باشند بازم مشکلی واس بچه وجود داره؟
سلام، با توجه به ژنوتیپ والدین و نوع توارثی که ممکن است در هنگام لقاح صورت بپذیرد، ممکن است فرزند سالم باشد یا به انواع آلفا تالاسمی مبتلا شود که هر یک در ضمن مطلب توضیح داده شده اند.
سلام وقت بخير
ببخشيد در آلفا تالاسمى جواب آزمايش الكتروفورز نرمال هست
و آيا ازدواج يك آلفا تالاسمى مينور با يك بتا تالاسمى مينور مشكل ساد هست براى بچه هاشون؟
سلام، خیر. اتفاقا ازدواج آلفا تالاسمی با بتا تالاسمی مینور موجب آلفابتا تالاسمی در کودکان میشود که هموگلوبین های نرمال تری نسبت به ازدواج دو آلفا تالاسمی و یا دو بتا تالاسمی دارند.